Parbati Biswas Group
Theory and Simulation Lab
Department of Chemistry
Dendritic Polymers
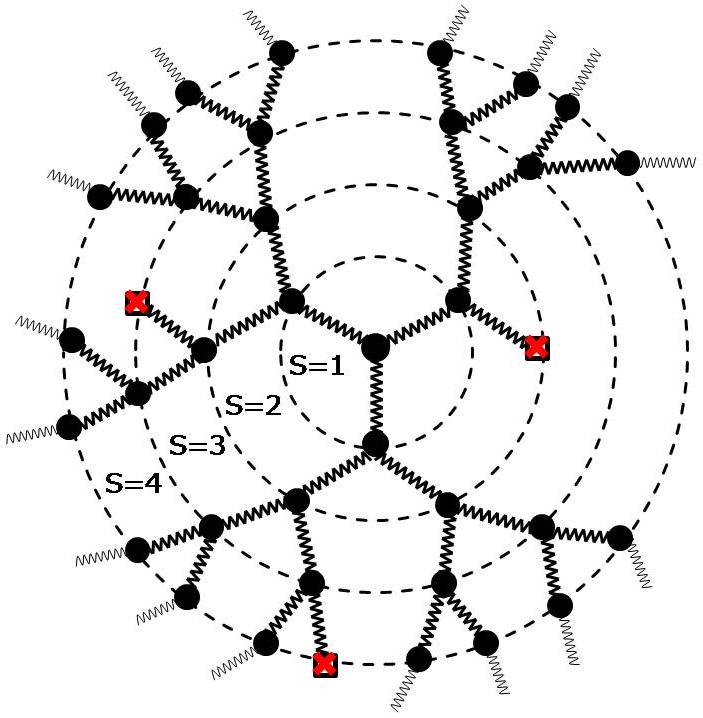
The hierarchical extensively branched architectures of dendrimers and randomly hyperbranched polymers have spurred considerable interest in exploring the novel polymer properties, both in bulk and solution. While dendrimers are highly branched symmetric, acyclic polymers centered on a multifunctional core resembling a Caylee tree, hyperbranched polymers represent imperfectly branched tree-like polymers with several scattered branch points throughout the molecule. Unlike dendrimers, hyperbranched polymers are devoid of a distinct core with well- defined generations of growth. A given number of monomers with a definite branching pattern correspond to only one dendrimer structure, while it may conform to a large number of hyperbranched structures because of different ways of distributing the branched and unbranched monomers. The conformational and dynamic properties of these polymers are explored in the framework of optimized Rouse-Zimm models. Various conformational and dynamic properties of dendrimers are investigated as a function of flexibility and/or excluded volume parameters and generational growth.
Intrinsically Disordered Proteins
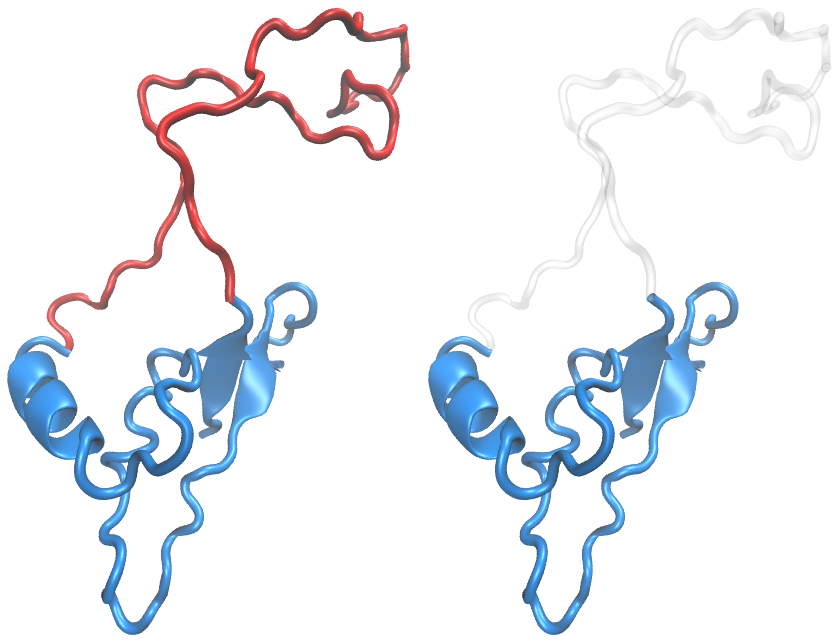
One of the outstanding interest of our group is to understand intrinsic disorder in proteins (IDPs), a functionally and structurally important class of proteins which perform diverse array of functions without a well-defined structure. We utilize theoretical and state-of-the-art computational techniques (molecular dynamics and Monte Carlo simulations) to have an idea about the amino acid sequence and conformational properties of IDPs and how these properties differ from that of the globular proteins. We have developed better prediction methods for IDPs based on the composition of amino acid triads in IDPs relative to that in globular proteins.
Hydration Water around Biomolecules
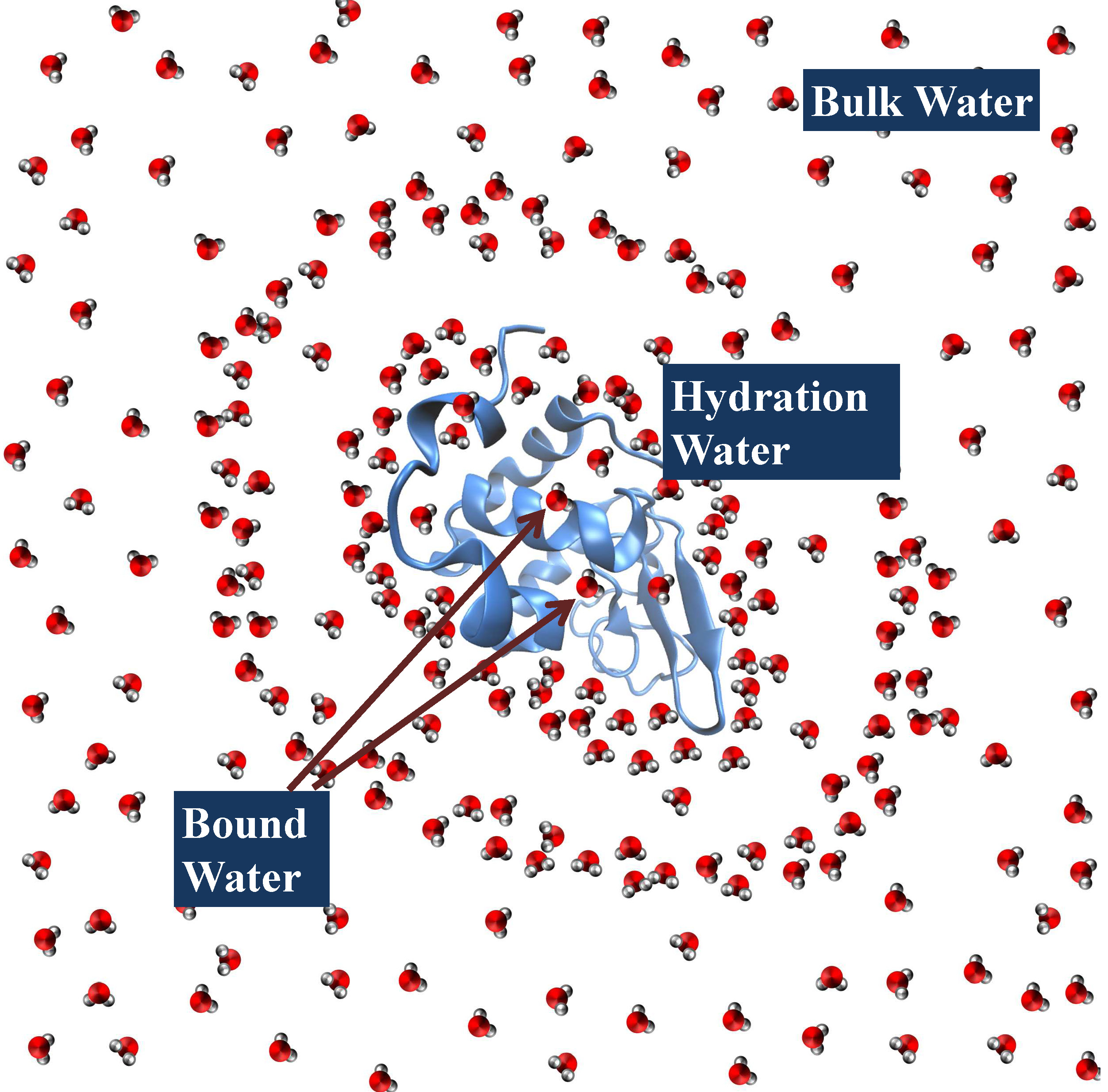
Another major area of work in our group includes the characterization of hydration water structure and dynamics around globular proteins, intrinsically disordered proteins and ambivalent helices. In this context, we have utilized the molecular dynamics simulations of proteins for the analysis of organization, structural arrangement, orientation and dynamics of hydration water around protein surface. In this regard, we have also investigated the effect of structural disorder on the hydrogen bonds between protein residues and hydration water molecules surrounding the protein surface.
Effect of the Environment on Proteins
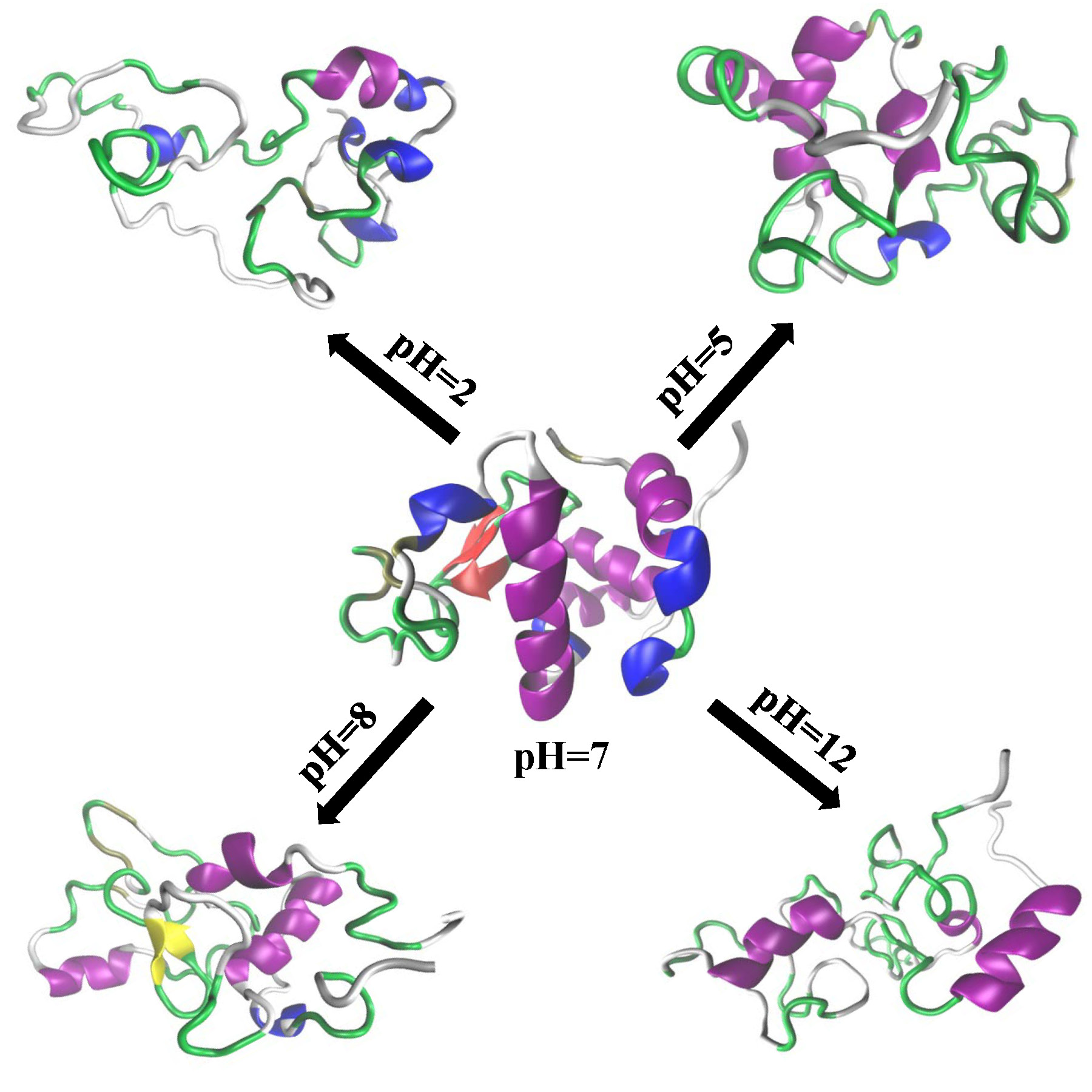
The effect of the environment
on the protein conformational properties constitutes another area of interest in
our group. We have used constant pH molecular dynamics simulations to analyze the
effect of changing pH on the structure of a globular protein (α-lactalbumin). Various
conformational properties e.g. radius of gyration, fractal dimension, root mean square
deviation, native contacts etc are studied to capture the impact of acidic and basic pH.
The change in the secondary structure content is also analyzed.
Protein Sequence Design
Designing sequences with funnel-like landscape, compatible with a predetermined target/native structure requires the energetic stabilization of the desired target/native structure against an ensemble of non-native/unfolded conformations. We use self-consistent mean field based methods to design sequences by optimizing an appropriate energy-based foldability criterion for a predetermined target structure. This method identifies the complete ensemble of sequences compatible with the target structure in terms of the monomer probabilities. We have utilized this method to better understand structural flexibility, effect of mutation, misfolding and fold switch in proteins.
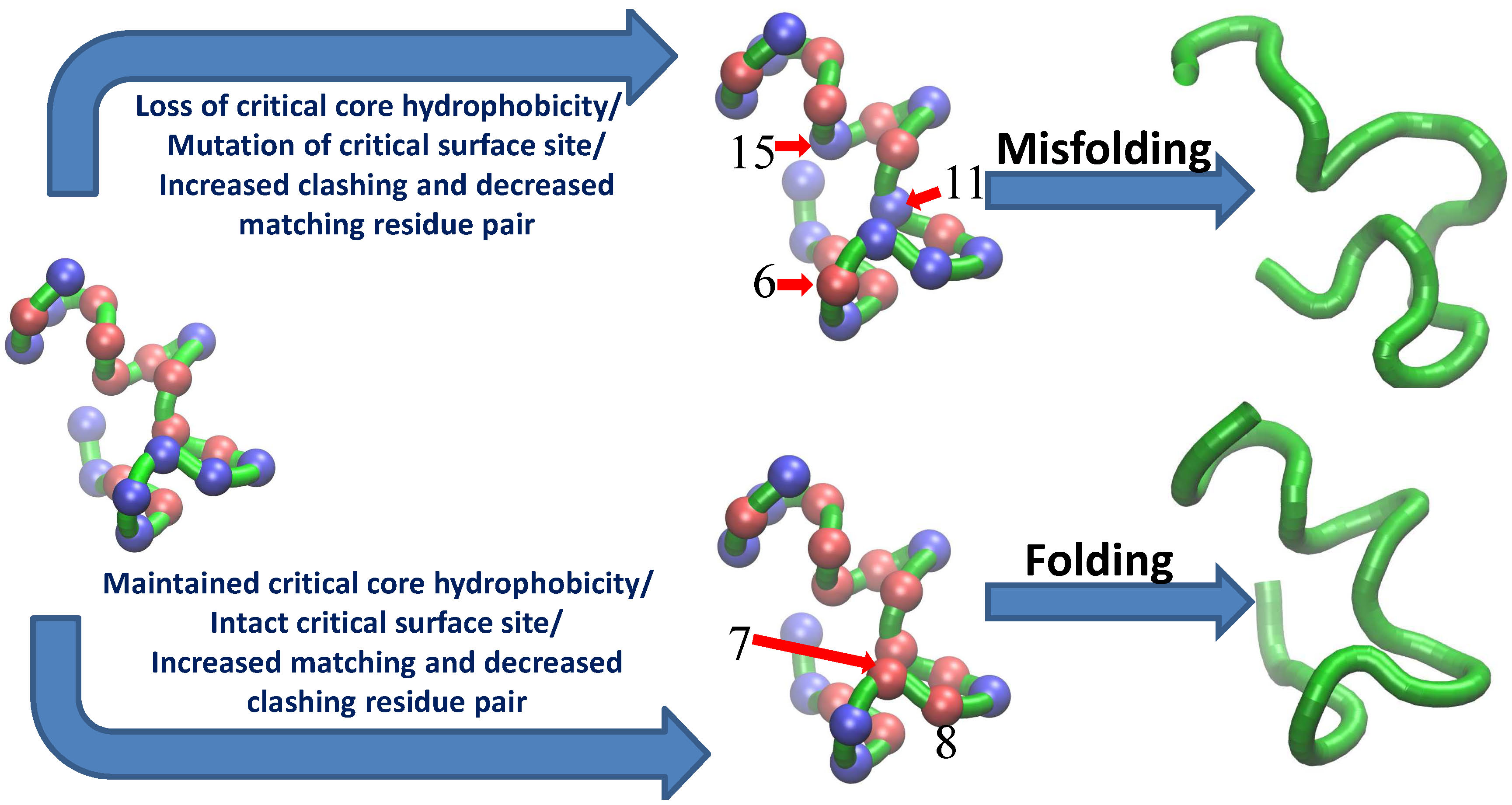
Protein Misfolding and Aggregation
Misfolding leads to the loss of
protein's function and promotes the formation of aggregates. These aggregates in many
cases organize to form 'amyloid fibrils'. All these conditions result
in many debilitating diseases in the organism. In our lab we are focused to
understand the underlying physics of misfolding and aggregation. We use various sequence
design methods and Monte Carlo simulation techniques to design and analyze the folded,
misfolded and unfolded sequences. The comparison between the three, provides important
insight into misfolding and aggregation.
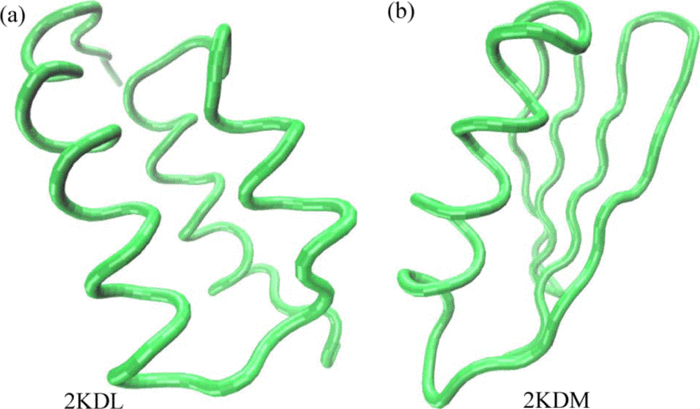
Protein Fold Switch
Designing "metamorphic"/"fold switch" proteins with multiple folds is challenging, since the design algorithm needs to optimize two different target structures simultaneously and ensure that there is a substantial overlap among them in the sequence space. We have used a self-consistent mean field based method to design pH induced fold switch sequences. We are interested to gain insight into the energy landscape and sequence characteristics of such fold switch sequences.
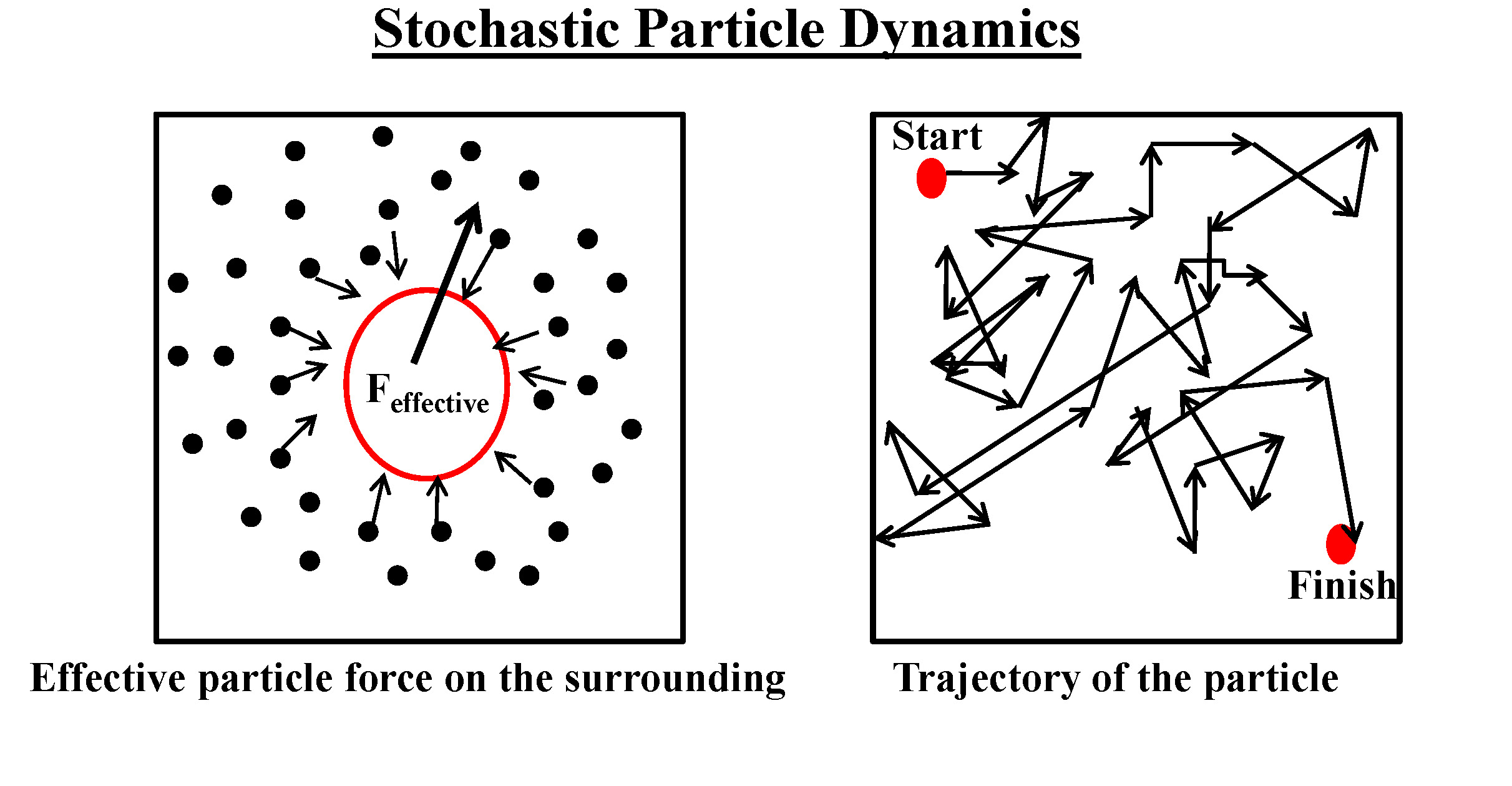
Stochastic Processes of Complex Dynamical Systems
We are interested in exploring variety
of problems ranging from the dynamical study of individual macromolecules
and mesoscopic physical properties of biological materials to the complex dynamics of the living cells
as a whole. In addition, our particular emphasis is on non-equilibrium processes, stochastic calculus,
chaotic theory, non-linear behaviour, non Fickian dynamics, boundary crossing problems and various
other interdisciplinary applications.
© Parbati Biswas